
Une découverte récente met en lumière un nouveau mode d'évolution des gènes associé au syndrome de Prader Willi
Dans les dernières décennies, les chercheurs ont réalisé que notre ADN produit non seulement des instructions pour fabriquer des protéines, mais aussi une multitude d'autres molécules appelées ARN non codants, essentielles pour son bon fonctionnement.
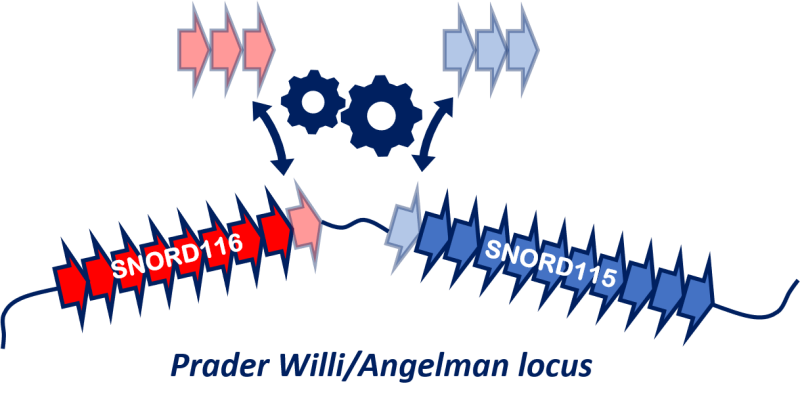
La dérégulation du nombre de ces ARN non codants est associée à des pathologies variées telles que le syndrome de Prader Willi, une maladie génétique rare dont les symptômes principaux sont une hypotonie et des difficultés de succion à la naissance puis, vers l’âge de 2 à 3 ans, un basculement vers une hyperphagie qui peut conduire à une obésité morbide si elle n’est pas strictement contrôlée. Les patients présentent également des anomalies endocriniennes et métaboliques ainsi que la propension à une déficience intellectuelle modérée. Le syndrome de Prader Willi implique très généralement l’absence d’expression de deux gènes produisant des ARN non codants nommés SNORD115 et SNORD116. Ces résultats de recherche ont fait l’objet d’une publication dans la prestigieuse revue NAR (Nuclear Acids Research) Molecular Medicine1 .
Les travaux en cours d'une équipe du laboratoire IMoPA ont permis de caractériser les premières cibles moléculaires des SNORD116 (Baldini et al. 2022) et de travailler à la réalisation d’outils thérapeutiques permettant de rétablir la fonction absente chez les patients (projet ASOPWS financé par la SATT Sayens). Contribuant à ces avancées, l’analyse comparative des dynamiques évolutives de tandems de gènes SNORD115 et SNORD116 chez les mammifères et dans les populations humaines a permis de préciser ces hypothèses à savoir que le problème principal réside dans l'absence d'expression du gène SNORD116, cause principale de la pathologie.
- 1Mathilde Guibert, Hélène Marty-Capelle, Anne Robert, Bruno Charpentier, Stéphane Labialle, Coordinated evolution of the SNORD115 and SNORD116 tandem repeats at the imprinted Prader–Willi/Angelman locus, NAR Molecular Medicine, Volume 1, Issue 1, January 2024, ugad003, https://doi.org/10.1093/narmme/ugad003
Ces recherches récentes ont permis d'identifier les premières cibles moléculaires de SNORD116 et de développer des outils thérapeutiques potentiels pour aider les patients.
Une découverte surprenante est que les deux ensembles de gènes SNORD115 et SNORD116 évoluent de manière coordonnée. Chez les mammifères, ils ont gagné ou perdu des copies ensemble au fil de l'évolution, et chez les grands singes, y compris les humains, les séquences de ces gènes se sont diversifiées pour former différentes sous-familles. Cette coordination semble liée à un mécanisme épigénétique particulier appelé empreinte génomique parentale, qui affecte le chromosome 15 de manière différente selon qu'il vient du père ou de la mère. Cette découverte suggère que ces gènes fonctionnent ensemble d'une manière qui nécessite un certain équilibre entre eux.
Cette hypothèse s’inscrit dans les travaux actuels de l’équipe visant à caractériser le répertoire complet des cibles moléculaires de ces gènes, ainsi qu’à affiner et tester le potentiel de molécules de thérapie du syndrome de Prader Willi. Ces résultats permettent de mieux comprendre le fonctionnement de ces gènes et ouvrent de nouvelles perspectives pour le développement de thérapies contre le syndrome de Prader Willi.
Source : Université de Lorraine